Membres de l'équipe
Réseaux de signalisation pour la stemness et la tumorigenèse
Notre équipe étudie la résilience versus la vulnérabilité des cellules aux fluctuations de signalisation pour comprendre comment le cancer se déclenche, évolue et répond aux traitements.
Notre équipe étudie la résilience versus la vulnérabilité des cellules aux fluctuations de signalisation pour comprendre comment le cancer se déclenche, évolue et répond aux traitements. Un outil clé de nos études est un cadre génétique unique chez la souris dans lequel une légère augmentation des niveaux de tyrosine kinase des récepteurs de type sauvage déclenche le programme tumorigène dans les tissus vulnérables : le foie et la glande mammaire. Ce cadre génétique “inside-out” est un modèle de cancer prédisposant ouvert à une variété d’altérations spontanées, récapitulant minutieusement les caractéristiques de la maladie survenant chez les patients humains. En combinant des outils complémentaires au sein d’approches interdisciplinaires, nous identifions de nouveaux mécanismes à l’origine de la déstabilisation de l’homéostasie tissulaire, de l’initiation et de la progression tumorale, ainsi que des vulnérabilités à exploiter pour des thérapies ciblées. De plus, nous étudions comment les changements de signalisation qualitatifs et quantitatifs conduisent à des comportements cellulaires diversifiés en utilisant des cultures de cellules souches, des organoïdes et des tumoroïdes, pour documenter comment la perception des signaux environnementaux et la modulation de l’intégrité épithéliale impactent le comportement cellulaire, l’interaction cellule-cellule et l’acquisition d’identités cellulaires diversifiées.
Publications
BCL-XL blockage in TNBC models confers vulnerability to inhibition of specific cell cycle regulators
BCL-XL blockage in TNBC models confers vulnerability to inhibition of specific cell cycle regulators
Strategies to Overcome Drug Resistance of Receptor Tyrosine Kinase-Addicted Cancer Cells.
Genetic analysis of specific and redundant roles for p38alpha and p38beta MAPKs during mouse development.
Actualités
Séminaire Interne par Olivier Castellanet
Une approche interdisciplinaire pour explorer et cibler la dialectique cancer-cellules immunitaires : d’un modèle de souris TNBC aux patients
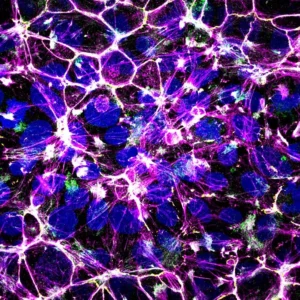
L’interaction entre la formation des jonctions serrées et les signaux liés à la gastrulation régule la différenciation du mésendoderme dans les hiPSCs
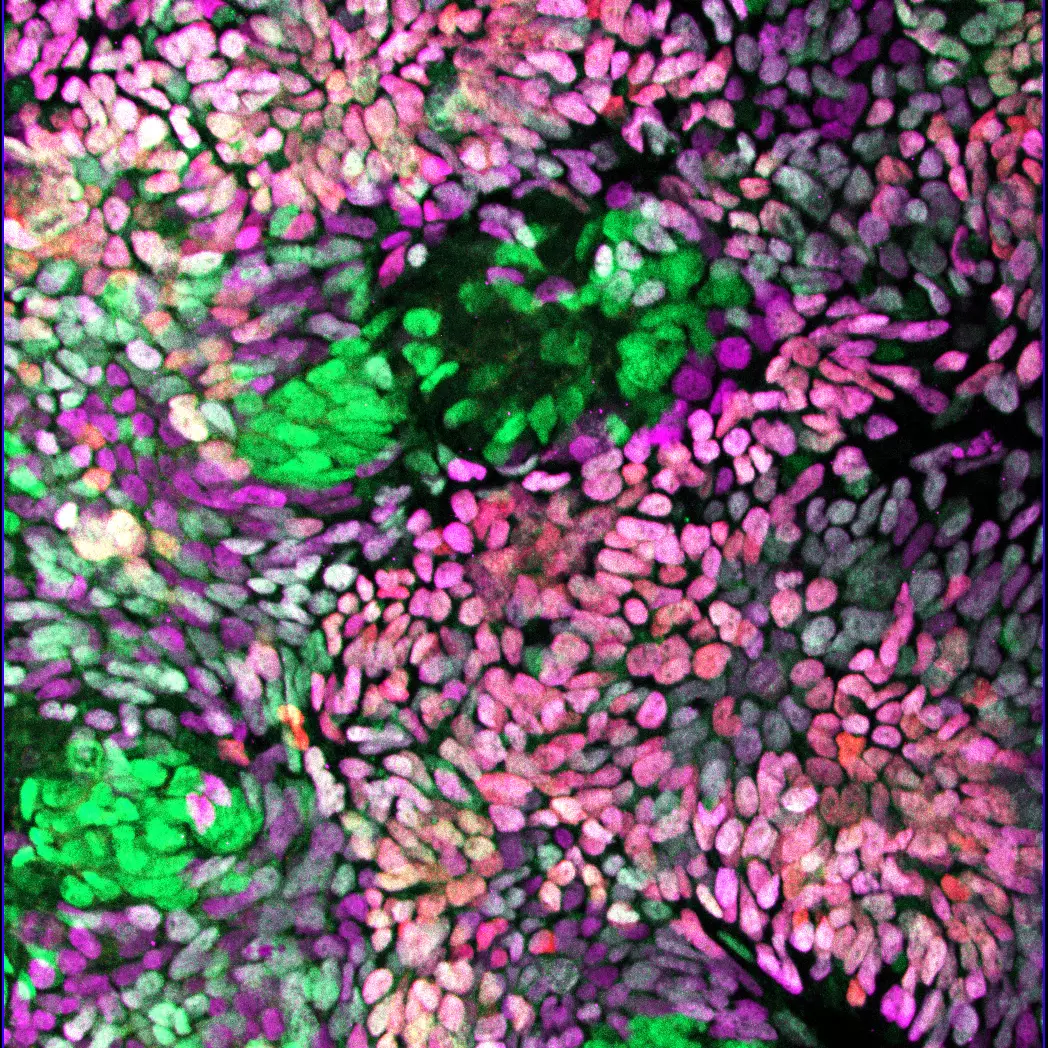
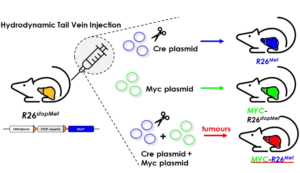
MYC et MET, les “Bonnie and Clyde” du foie
Une nouvelle coopération génique agissant sur le cancer du foie a été découverte chez des patients et documentée fonctionnellement in vivo
Félicitation à Aziz Moqrich, Sophie Chauvet et Stefan Harmansa.

L’équipe de Flavio Maina publie, dans la revue Theranostics, des travaux qui illustrent comment la spécificité du ciblage des régulateurs du cycle cellulaire est déterminante pour la mise au point de thérapies anticancéreuses combinatoires.
The teams of F Maina and L Kerkerian-Le Goff publish in Stem Cells Translational Medicine a work led by Rosanna Dono showing that levels of the signal modulator GLYPICAN-4 are critical for the generation of midbrain dopamine neurons from human induced pluripotent stem cells and for overcoming their tumorigenic properties.
L’équipe Maina publie dans Advanced Science la mise en place d’un système génétique modélisant, chez la souris, l’hétérogénéité et la résistance aux traitements du cancer du sein dit Triple-Négatif humain (TNBC), un type de cancer du sein très agressif et pour lequel il n’y a pas de traitement efficace à l’heure actuelle.
L’équipe a découvert que ADAMTSL5 est un oncogène épigénétiquement activé et surexprimé chez une grande partie des patients humains atteints de HCC et dans des modèles murins de HCC.
Les pixels du CERN ont permis de suivre la croissance de tumeurs spontanées chez la souris.
Mission Contribuer aux recherches de l’équipe sur la caractérisation fonctionnelle etmécanistique des circuits de signalisation dans le cancer. Développer de nouveaux outilsthérapeutiques et diagnostiques. Activités
Membres de l'équipe
Alumni
Les organismes qui nous financent



{kind=link}