Team members
Signalling networks for stemness and tumorigenesis
Our team studies resilience versus vulnerability of cells to signalling fluctuations to understand how cancer starts, evolves, and responds to treatments.
Our team studies resilience versus vulnerability of cells to signalling fluctuations to understand how cancer starts, evolves, and responds to treatments. A key tool in our studies is a unique mouse genetic setting in which slightly increased wild-type receptor tyrosine kinase levels trigger the tumorigenic program in vulnerable tissues: liver and mammary gland. This “inside-out” genetic setting is a predisposing cancer model open to a variety of spontaneous alterations, thoroughly recapitulating disease features occurring in human patients. By combining complementary tools within interdisciplinary approaches, we identify new mechanisms at the root of tissue homeostasis destabilisation, tumour initiation, progression, as well as vulnerabilities to exploit for targeted therapies. Furthermore, we study how qualitative and quantitative signalling changes lead to diversified cell behaviours using stem cell cultures, organoids, and tumoroids, to document how perception of environmental signals and modulation of epithelial integrity impact cell behaviour, cell-cell interaction, and the acquisition of diversified cell identities.
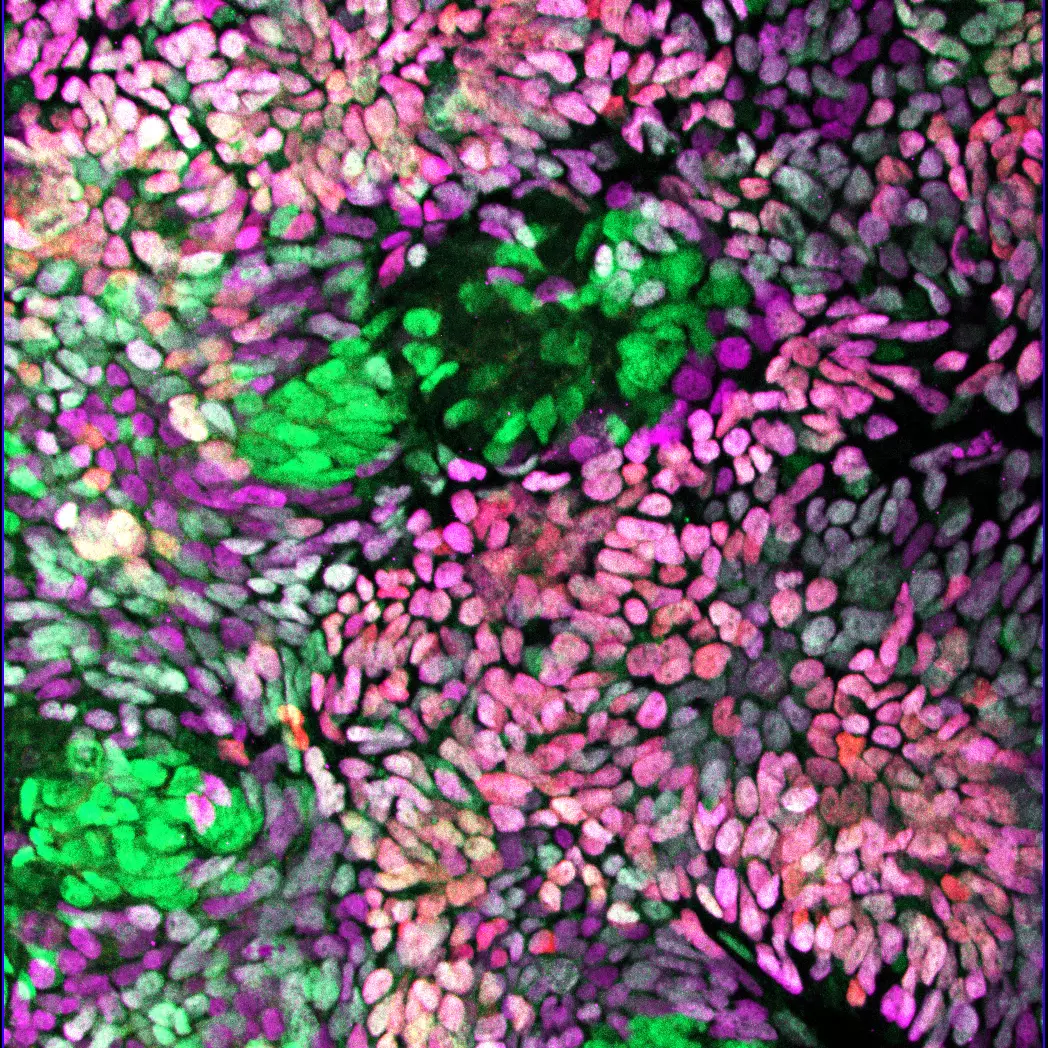
Publications
BCL-XL blockage in TNBC models confers vulnerability to inhibition of specific cell cycle regulators
BCL-XL blockage in TNBC models confers vulnerability to inhibition of specific cell cycle regulators
Strategies to Overcome Drug Resistance of Receptor Tyrosine Kinase-Addicted Cancer Cells.
Genetic analysis of specific and redundant roles for p38alpha and p38beta MAPKs during mouse development.
News
Internal Seminar by Olivier Castellanet
An interdisciplinary approach to explore and target the cancer-immune cell crosstalk: from a TNBC mouse model to patients
How disruption drives change
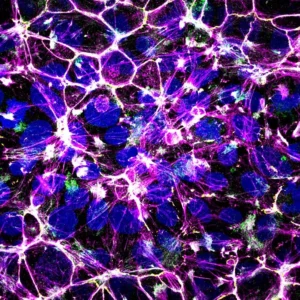
Interaction between tight junction formation and gastrulation signals regulates mesendoderm differentiation in hiPSCs
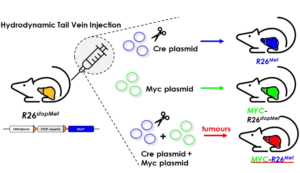
A new genetic cooperation acting on liver cancer has been uncovered in patients and functionally documented in vivo
Through an international collaboration, using breast cancer as a biological paradigm, Flavio Maina Team publishes a work in the Theranostics journal exemplifying how specificity in targeting cell cycle regulators is essential for combinatorial cancer therapies.
The teams of F Maina and L Kerkerian-Le Goff publish in Stem Cells Translational Medicine a work led by Rosanna Dono showing that levels of the signal modulator GLYPICAN-4 are critical for the generation of midbrain dopamine neurons from human induced pluripotent stem cells and for overcoming their tumorigenic properties.
Maina Team publishes in Advanced Science the setup of a genetic system that models, in mice, the heterogeneity and primary resistance to treatment of human Triple Negative Breast Cancer (TNBC).
Maina Team publishes in Journal of Hepatology. The team found that ADAMTSL5 is an epigenetically activated oncogene overexpressed in a large fraction of human HCC patients and in HCC mouse models.
Researchers have used the CERN pixels to follow tumour evolution in a liver cancer mouse model.
The mission is to contribute to the team research on mechanistic and functional characterization of signalling circuits in cancer, develop novel therapeutic and diagnostic tools.
Team members


Alumni
Funding bodies



{kind=link}