Team members
Cellular Interactions, Neurodegeneration and Neuroplasticity
We investigate the organization and functional dynamics of brain neuronal circuits, their adaptive capacities and the mechanisms underlying their pathological dysfunction and neurodegeneration, with focus onto the basal ganglia network.
Our main research interest is on synaptic transmission, neurodegeneration and neuroplasticity in the adult brain. Communication between neurons at the level of their connections, called synapses, is the substrate of information processing in the networks underlying brain functions. Synaptic transmission is a highly dynamic and regulated process, influenced by glial cells, whose abnormalities are associated with a number of brain diseases (concept of synaptopathies). Neurodegeneration is a pathological process that triggers progressive dysfunction and death of nerve cells. Understanding the pathological mechanisms triggering and sustaining neurodegeneration (pathogenesis), its consequences on circuit function and the mechanisms that help neurons to manage cellular stress is essential for the development of curative or disease-modifying treatments for devastating neurodegenerative disorders, such as Parkinson’s disease (PD). Neuroplasticity refers to the capacity of the nervous system to adapt in response to experience and to internal or external stimulations by modifying the interactions between nerve cells, including changes in the number of synapses and the efficacy/strength of synaptic transmission (synaptic plasticity), or by generating new nerve cells. This faculty is not limited to development, but occurs lifelong, although declining with aging. Neuroplasticity has been notably involved in learning and memory processes. It also occurs under pathological conditions or in response to chronic treatments. Such adaptive changes can represent compensatory mechanisms counteracting the deficits triggered by neuronal dysfunction or death, delaying the symptom onset, or, on the contrary, can participate in or even aggravate the deficits.
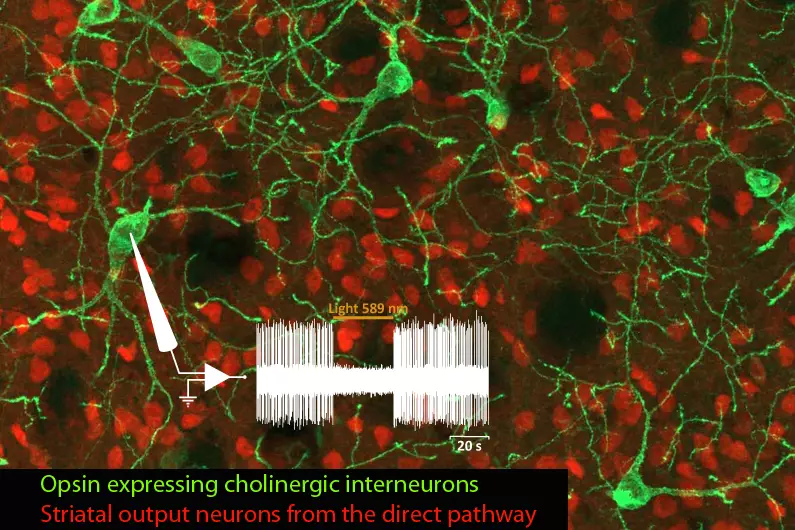
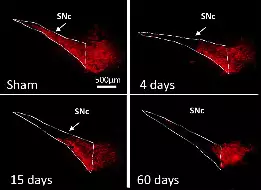
The team is investigating these processes in the context of basal ganglia (BG)-related functions and pathologies, in particular PD, a movement disorder characterized by the degeneration of midbrain dopamine neurons innervating the striatum, the main BG input station. Through collaborations, our work also addresses fundamental and clinically-relevant issues in the context of other neuropathologies, including autism spectrum disorder (ASD), Alzheimer’s disease and Charcot-Marie-Tooth disease.
Publications
Glutamate transporter 1-expressing glia in the rat substantia nigra-Morphometric analysis and relationships to synapses
Processing of information from the parafascicular nucleus of the thalamus through the basal ganglia
Glutamate transporter 1-expressing glia in the rat substantia nigra-Morphometric analysis and relationships to synapses
Deep brain stimulation mechanisms: beyond the concept of local functional inhibition.
[Acetylcholinesterase inhibitors in Alzheimer’s disease: further comments on their mechanisms of action and therapeutic consequences].
Effects of GPi and STN inactivation on physiological, motor, cognitive and motivational processes in animal models of Parkinson’s disease.
Rationale for targeting the thalamic centre-median parafascicular complex in the surgical treatment of Parkinson’s disease.
Impact of surgery targeting the caudal intralaminar thalamic nuclei on the pathophysiological functioning of basal ganglia in a rat model of Parkinson’s disease.
Metabotropic glutamate 5 receptor blockade alleviates akinesia by normalizing activity of selective basal-ganglia structures in parkinsonian rats.
News
3 motivated and talented students successfully defended their thesis between February and July 2023.

5 motivated and talented students successfully defended their thesis between September 2022 and January 2023.
Lydia Kerkerian-Le Goff, Research Director at the CNRS and head of the Cellular Interactions, Neurodegeneration and Neuroplasticity team at the Marseille Institute of Developmental Biology (IBDM)

This study published in Cell Death & Disease, conducted by the team of L. Kerkerian-Le Goff provides the first evidence for a role of the stress-induced protein TP53INP1 in the maintenance of neuronal homeostasis in ageing and Parkinson’s disease-related stress conditions.
No jobs opportunities found..
Team members


Alumni
Funding bodies