Membres de l'équipe
Interactions cellulaires, neurodégénérescence et neuroplasticité
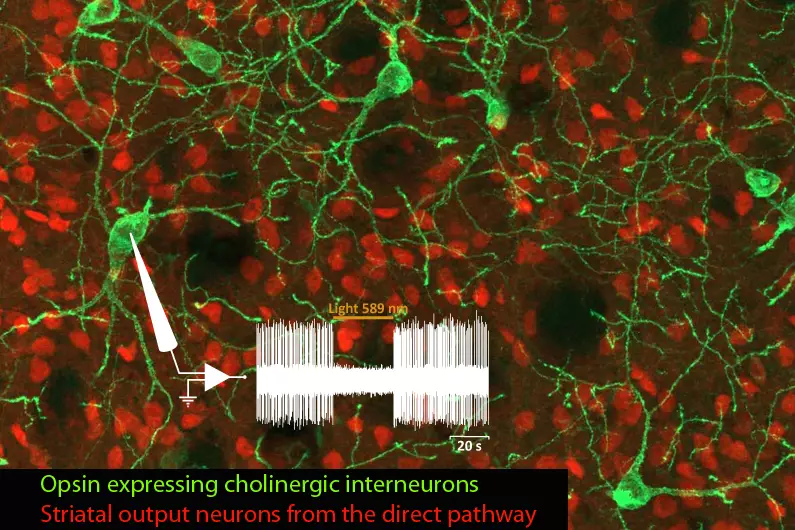
Nous étudions l'organisation et la dynamique fonctionnelle des circuits neuronaux du cerveau, leurs capacités d'adaptation et les mécanismes qui sous-tendent leur dysfonctionnement pathologique et leur neurodégénérescence, avec un focus sur le réseau des ganglions de la base.
Notre principal intérêt de recherche porte sur la transmission synaptique, la neurodégénération et la neuroplasticité dans le cerveau adulte. La communication entre les neurones au niveau de leurs connexions, appelées synapses, est le substrat du traitement de l’information dans les réseaux qui sous-tendent les fonctions cérébrales. La transmission synaptique est un processus hautement dynamique et régulé, influencé par les cellules gliales, dont les anomalies sont associées à un certain nombre de maladies cérébrales (notion de synaptopathies). Neurodégénérescence est un processus pathologique qui déclenche le dysfonctionnement et la mort progressive des cellules nerveuses. La compréhension des mécanismes pathologiques qui déclenchent et entretiennent la neurodégénérescence (pathogenèse), de ses conséquences sur le fonctionnement des circuits et des mécanismes qui aident les neurones à gérer le stress cellulaire est essentielle pour le développement de traitements curatifs ou modificateurs de la maladie pour les troubles neurodégénératifs dévastateurs, tels que la maladie de Parkinson (MP). Neuroplasticité. désigne la capacité du système nerveux à s’adapter en réponse à l’expérience et aux stimulations internes ou externes en modifiant les interactions entre les cellules nerveuses, y compris les changements dans le nombre de synapses et l’efficacité/la force de la transmission synaptique (plasticité synaptique), ou en générant de nouvelles cellules nerveuses. Cette faculté n’est pas limitée au développement, mais se produit tout au long de la vie, bien qu’elle diminue avec le vieillissement. La neuroplasticité a notamment été impliquée dans les processus d’apprentissage et de mémoire. Elle se produit également dans des conditions pathologiques ou en réponse à des traitements chroniques. Ces changements adaptatifs peuvent représenter des mécanismes compensatoires contrecarrant les déficits déclenchés par le dysfonctionnement ou la mort neuronale, retardant l’apparition des symptômes, ou, au contraire, participer à ces déficits, voire les aggraver.
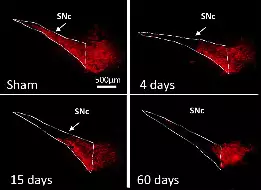
L’équipe étudie ces processus dans le contexte des fonctions et des pathologies liées aux ganglions de la base (BG), en particulier la MP, un trouble du mouvement caractérisé par la dégénérescence des neurones dopaminergiques du mésencéphale innervant le striatum, la principale station d’entrée des BG. Par le biais de collaborations, nos travaux abordent également des questions fondamentales et cliniquement pertinentes dans le contexte d’autres neuropathologies, notamment les troubles du spectre autistique (TSA), la maladie d’Alzheimer et la maladie de Charcot-Marie-Tooth.
Publications
Glutamate transporter 1-expressing glia in the rat substantia nigra-Morphometric analysis and relationships to synapses
Processing of information from the parafascicular nucleus of the thalamus through the basal ganglia
Glutamate transporter 1-expressing glia in the rat substantia nigra-Morphometric analysis and relationships to synapses
Deep brain stimulation mechanisms: beyond the concept of local functional inhibition.
[Acetylcholinesterase inhibitors in Alzheimer’s disease: further comments on their mechanisms of action and therapeutic consequences].
Effects of GPi and STN inactivation on physiological, motor, cognitive and motivational processes in animal models of Parkinson’s disease.
Rationale for targeting the thalamic centre-median parafascicular complex in the surgical treatment of Parkinson’s disease.
Impact of surgery targeting the caudal intralaminar thalamic nuclei on the pathophysiological functioning of basal ganglia in a rat model of Parkinson’s disease.
Metabotropic glutamate 5 receptor blockade alleviates akinesia by normalizing activity of selective basal-ganglia structures in parkinsonian rats.
Actualités
3 étudiants ont soutenu leur thèse avec succès entre février et juillet 2023.

Lydia Kerkerian-Le Goff, Research Director at the CNRS and head of the Cellular Interactions, Neurodegeneration and Neuroplasticity team at the Marseille Institute of Developmental Biology (IBDM)

L’étude menée par l’équipe de L. Kerkerian-Le Goff apporte les premières évidences en faveur d’un rôle de la protéine induite par le stress TP53INP1 dans le maintien de l’homéostasie neuronale en condition de stress lié au vieillissement normal et à la Maladie de Parkinson (MP).
No jobs opportunities found..
Membres de l'équipe
Alumni
Les organismes qui nous financent